Reconstructing the Shattered Niche: Mechanical Strain as a Catalyst for In Vitro Traumatic Brain Injury
Traumatic brain injury (TBI) fundamentally originates from mechanical impact, yet conventional neuroscience heavily relies on rigid, two-dimensional platforms that sentence neurons to a physics-deprived silence. This methodological blind spot fails to capture the post-traumatic phenotype, severely limiting the clinical translation of therapeutic candidates. By engineering a kinetic disease-modeling platform that applies a controlled, disruptive 25% uniaxial cyclic stretch at 1 Hz to human neuroblastoma (SH-SY5Y) cells on compliant PDMS membranes, this approach abandons the quest for optimized growth. Instead, it redefines the parameters of disease modeling, elevating both human-relevance and spatiotemporal precision to recapitulate the neurodegenerative microenvironment in vitro.
The Biomechanical Cascade:
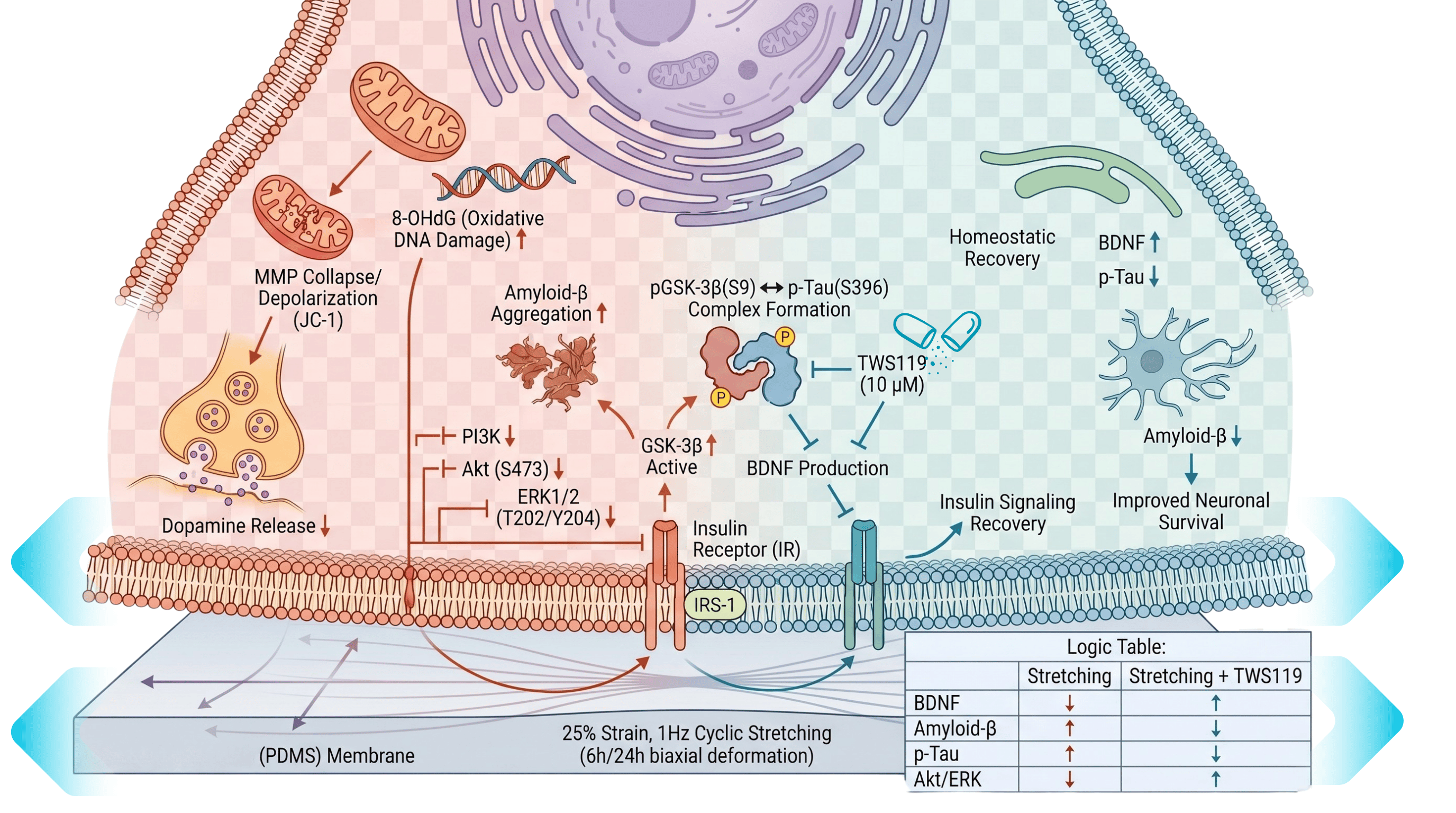
From Physical Impact to Metabolic Sabotage Subjecting human neuroblasts to this violent physical deformation for 6 to 24 hours does not merely stretch the cell; it dismantles neuronal homeostasis. The 25% strain collapses the mitochondrial membrane potential (MMP), triggering a surge in oxidative DNA damage marked by elevated 8-OHdG. At the neurochemical level, this mechanical insult stifles the synthesis of dopamine and brain-derived neurotrophic factor (BDNF). More tellingly, the physical stress forces the pathological accumulation of amyloid-beta (Aβ) peptides and hyperphosphorylates Tau proteins—reproducing the hallmark secondary neurodegenerative sequelae traditionally thought to be unachievable in isolated culture.
Overcoming the Static Phenotype:
The Deficient Control The transition from static confinement to biomimetic dynamics unmasks the signaling networks hijacked by physical trauma. While static control cells maintain physiological BDNF secretion and undisturbed insulin signaling, the introduction of the 25% cyclic tension forces cellular realignment perpendicular to the axis of stress. This structural deformation abruptly dampens the neuroprotective PI3K/Akt and ERK survival cascades, culminating in the aberrant activation of glycogen synthase kinase-3 beta (GSK-3β). Once unleashed, GSK-3β drives the assembly of the GSK-3β-Tau complex, executing axonal injury and protein aggregation that static cultures simply cannot spontaneously generate. Dynamic mechanical force thus emerges not merely as a stimulus, but as a mandatory prerequisite for triggering secondary neurodegeneration.
Transcending the Rodent Black Box:
Species-Independent Resolution Historically, TBI research has surrendered to the systemic noise of rodent models, which are frequently obscured by uncontrolled inflammatory responses, heterogeneous injury distribution, and evolutionary divergence. This in vitro stretching platform bypasses these limitations by deploying human SH-SY5Y neurons that preserve essential dopamine-β-hydroxylase activity. By stripping a chaotic cranial trauma down to its pure physical coordinates (25% strain, 1 Hz), this system isolates mechanical force from systemic endocrine variables. This unprecedented resolution allowed the definitive validation of the GSK-3β inhibitor TWS119 in halting mechanotransduction-mediated degeneration—offering a clear mechanistic readout that live animal models routinely shroud in systemic complexity.
A static petri dish merely permits neurons to subsist; only through pathologically calibrated, biomimetic pulsation do human cells betray the hidden molecular mechanics of traumatic degeneration.